Kromozom 3 (insan)
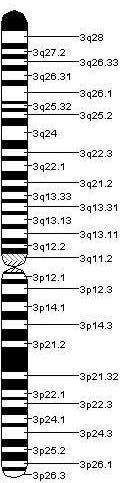
Kromozom 3; 22 çift otozomal insan kromozomlarından 3. olanıdır. İnsanlarda normalde bir çift halinde bulunur. 200 milyon baz çiftine ve toplam hücre DNA'sının %6,5'ine sahiptir. Kromozom 3 muhtemelen 1,100 ile 1,500 arasında gen içermektedir.
İnsan karyotipinde kromozomlar
Genler
Kromozom 3'de bulunan genlerden bazıları şunlardır;
- ALAS1: aminolevulinat, delta-, sentetaz 1
- BTD: biotinidaz
- CCR5: Kemokin (C-C motif) resepör 5
- CPOX: koproporfrinojen oksidaz (koproporfriya, harderoporfriya)
- HGD: homojentisat 1,2-dioksigenaz (homojentizat oksidaz)
- MCCC1: metilkrotonoyl-Koenzim A karboksilaz 1 (alfa)
- MITF: mikroftalmia-associated transkripsiyon faktörü
- MLH1: mutL homolog 1, kolon kanseri, poliptik olmayan tip 2 (E. coli)
- PCCB: propionil koenzim A karboksilaz, beta polipeptid
- PDCD10: programlanmış hücre ölümü 10
- PIK3CA: fosfoinositid 3-kinaz, katalitik, alfa polipeptid
- RAB7: RAB7, üye RAS onkogen ailesi
- SCN5A: sodyum kanalı, voltaj kapısı, tip V, alfa (uzun QT sendromu 3)
- SLC25A20: çözünebilir taşıyıcı ailesi 25 (karnitin/asilkarnitin translokaz), üye 20
- TMIE: geçiş zarı iç kulak
- USH3A: Usher sendromu 3A
- VHL: von Hippel-Lindau tümör baskılayıcı
- ZNF9: çinko parmak protein 9
Hastalıklar
Kromozom 3 üzerinde bulunan genlerin yol açtığı hastalıklardan bazıları şöyledir;
- alkaptonüri
- biotinidaz eksikliği
- Brugada sendromu
- karnitin-asilkartinin translokaz eksikliği
- serebral derin malformasyon
- Charcot-Marie-Tooth hastalığı
- Charcot-Marie-Tooth hastalığı, tip 2
- hereditai koproporfriya
- hereditari nonpolypozis kolorektal kanser]
- 3-metilkrotonil-CoA karboksikaz eksikliği
- miyotonik distrofi
- miyotonik distrofi, tip 2
- sendromik olmayan sağırlık
- sendromik olmayan sağırlık, otozomal çekinik
- poryfriya
- propionik asidemi
- Romano-Ward sendromu
- Usher sendromu
- Usher syndrome tip III
- von Hippel-Lindau sendromu
- Waardenburg sendromu
- Otizim
- Gece körlüğü
- Sağırlık
- HIV enfeksiyonu, duyarlı/dayanıklı
- Diabet
- Göğüs/kolon/akciğer/pankreas kanseri
- Usher sendromu/ Usher sendromu (Finlandiya)
- Yumurtalık kanseri
- Muir-Torre ailevi kanser sendromu
- Glokom, ilk açık açı
- Essensial titretme
- Kısa boy
- Lökoensefalopati
- Harderoporfirinuria
- Spinoserebellar ataksia
- Moyamoya hastalığı
- Endplate asetlikolinesteraz eksikliği
- Hypobetalipoproteinemia, ailesi
- Septooptic dysplasia
- Uzun QT sendromu
- Katarakt
- Mukopolisakkaridozis
- Kalp tıkanması, aşamalı/aşamasız
- Malign hipertermia hassasiyeti
- Atransferrinemia
- Sükroz intolerans
- Serebral cavernous malformasyonu
- Nöropati, ırsi motor ve duyumsal, Okinawa tip
- Protein S yetmezliği
- T-hücresi lösemi translokasyon geç dönem geni
- Kseroderma pigmentosum, tamamlayıcı grup c
- Hailey-Hailey hastalığı
- Koproporfyria
- Dopamin reseptörü
- Glikojen depolama hastalığı
- Arritmojenik sağ ventriküler displazi
- Pseudo-Zellweger sendromu
- Blefarofimozis, epicantus inversus and ptosis type 1
- Lenfoma
- Metaplazel kondrodisplazi, Murk Jansen tipi
- Moebius sendromu
- Epidermolizis bullosa
Düzensizlikleri
- 3p kısmi trizomisi, Kranio-fasial düzensizlikler ve mental gerilik ve düğümlü parmak izi karakteristiktir.
- Duplikasyon-delesyon sendromu, uzun kol ucu duplikasyonu ve kısa kol ucu delesyonları da prenatal ve postnatal gelişme geriliğ, genital organ düzensizlikleri, mental gerilik, konjenital kalp rahatsızlıklarına nedendir. Mayotik nondisjunctiona bağlı ortaya çıkmaktadır.
| ||||||||||
This article is issued from Vikipedi - version of the 1/23/2016. The text is available under the Creative Commons Attribution/Share Alike but additional terms may apply for the media files.